Manifestazioni cutaneo-mucose di malattie autoimmuni
Coordinamento a cura di Edoardo Cataudella e Fabiana Furci
Lupus Eritematoso Cutaneo – Sintomatologia, decorso e presentazioni cliniche
Il lupus eritematoso cutaneo (LEC) rappresenta uno spettro eterogeneo di manifestazioni dermatologiche che rientrano nell’ambito delle malattie autoimmuni.
Si tratta di un gruppo di dermatosi mediate da un’alterazione dell’immunità innata e adattativa con spiccato fototropismo, in cui l’interazione tra predisposizione genetica, esposizione ai raggi UV, farmaci e altri fattori ambientali contribuisce all’attivazione aberrante dell’immunità e al danno tissutale, mediato principalmente dagli interferoni del tipo I (IFNs tipo I) e dall’infiltrato linfomonocitario (dermatite di interfaccia). Il quadro clinico è ampio e comprende forme acute, subacute e croniche, come definito nelle classificazioni correnti.
Decorso
Le manifestazioni cutanee del lupus mostrano un andamento cronico con riacutizzazioni spesso legate a esposizione solare, stress, infezioni, variazioni climatiche o farmaci fotosensibilizzanti. L’interessamento di aree fotoesposte rappresenta un tratto distintivo comune, riflettendo il ruolo centrale dei trigger ambientali nella patogenesi.
La relazione con il lupus sistemico varia in base al sottotipo:
- l’ACLE è più frequentemente associato a lupus sistemico conclamato;
- lo SCLE presenta un rischio intermedio e variabile;
- le forme croniche, ad eccezione del DLE disseminato, evolvono raramente verso forme sistemiche;
- la variante intermittente/tumida (ICLE) non presenta rischio significativo di progressione.
Diagnosi differenziale
Il lupus cutaneo presenta aspetti molto polimorfi, sovrapponibili a numerose dermatosi infiammatorie o fotoindotte. La diagnosi differenziale può includere rosacea, dermatite seborroica, psoriasi, dermatomiosite, lichen planus, fotodermatiti, infezioni fungine o virali, oltre a reazioni da farmaco. In questo contesto l’istologia svolge un ruolo fondamentale: il riscontro tipico è quello di una dermatite di interfaccia (lichenoide), caratteristica ma non esclusiva del lupus, utile nel supportare la diagnosi insieme ai dati clinici e immunologici.
Terapia
La gestione del lupus cutaneo si basa sulla combinazione di misure di fotoprotezione rigorosa, terapia topica e trattamento sistemico nei casi più estesi o refrattari.
- Topici:corticosteroidi a diversa potenza e, in casi selezionati, dapsone topico possono aiutare nel controllo delle lesioni attive.
- Sistemici:corticosteroidi, ciclosporina o voclosporina, idrossiclorochina e altri antimalarici rimangono cardini terapeutici. Nei casi resistenti si può ricorrere a retinoidi, DMARD convenzionali (azatioprina, metotrexato, micofenolato mofetile) e, nelle forme più severe o refrattarie, a terapie biologiche come belimumab, rituximab o anifrolumab, che hanno progressivamente ampliato l’armamentario terapeutico del lupus cutaneo.
Forme acute (ACLE)
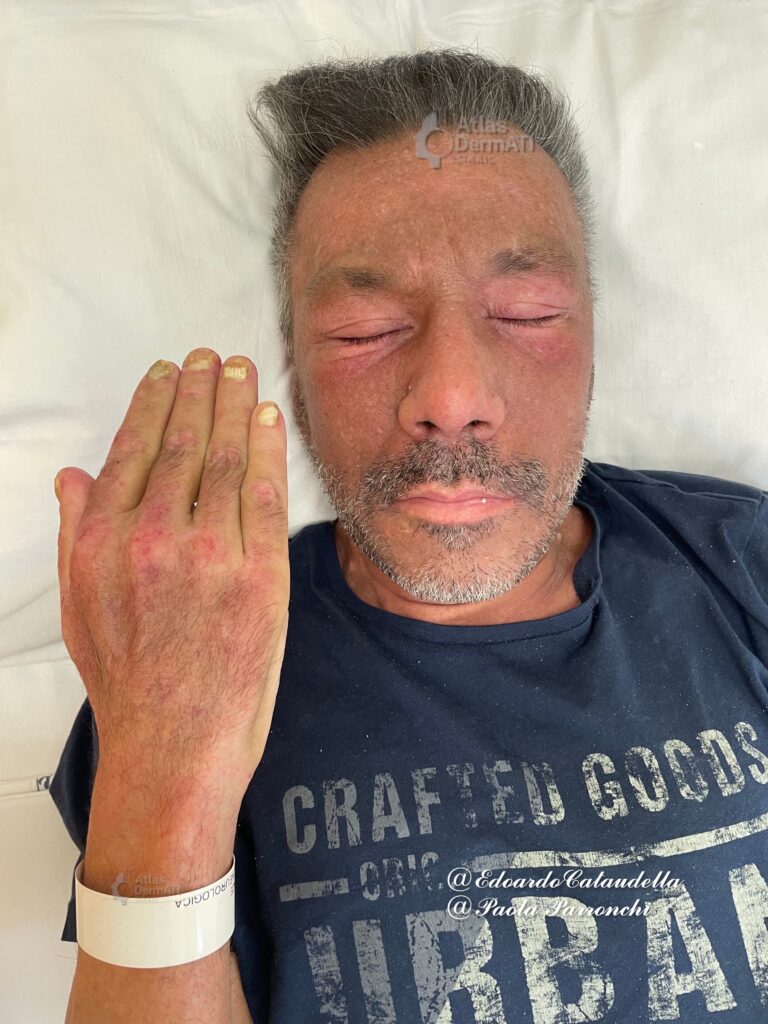
Nelle forme acute l’esordio è spesso brusco e correlato all’attività sistemica della malattia. La manifestazione più tipica è l’eritema malare a distribuzione “a farfalla”, caratterizzato da chiarezza della regione naso-labiale e netta fotosensibilità. Può presentarsi anche un esantema maculo-papulare diffuso. Le lesioni sono generalmente non cicatriziali e tendono a seguire l’andamento dell’attività sistemica. L’associazione con sintomi sistemici è comune: febbre, astenia, artralgie e mialgie, calo ponderale e sudorazioni possono accompagnare il quadro cutaneo, riflettendo un coinvolgimento multisistemico più ampio.
Forme subacute (SCLE)
Il lupus eritematoso subacuto è caratterizzato da lesioni annulari policicliche oppure da chiazze papulo-squamose a morfologia psoriasiforme, spesso distribuite su superfici fotoesposte. L’esordio è più graduale rispetto alle forme acute. Le lesioni possono essere altamente sensibili alla luce ultravioletta e raramente esitano in cicatrici, pur potendo lasciare alterazioni pigmentarie persistenti. Il rischio di progressione verso una forma sistemica è presente ma modesto e molto variabile, e tende a dipendere da fattori quali autoanticorpi specifici, durata della malattia e presenza di segni costituzionali.
Forme croniche (CCLE)
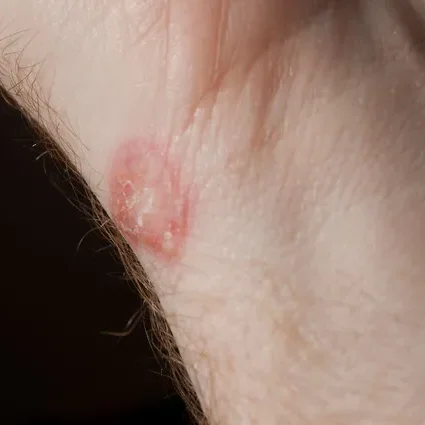
Il lupus eritematoso cutaneo cronico comprende un ventaglio di varianti che condividono un decorso protratto e una tendenza alla cicatrizzazione:
- Lupus discoide (DLE), localizzato o disseminato, caratterizzato da placche eritemato-ipoeritematose, follicolocentriche, con ipercheratosi aderente, telangiectasie, atrofia e cicatrici permanenti. La localizzazione più comune è il volto e il cuoio capelluto, con possibile alopecia cicatriziale.
- Panniculite lupica (lupus profundus), con noduli o placche profonde, dolenti e spesso seguite da lipoatrofia residua.
- Lupus geloni (chilblain lupus), tipicamente indotto dal freddo, con lesioni violacee o eritemato-edematose su mani, piedi, orecchie.
- Lupus eritematoso tumido (LET/ICLE), caratterizzato da lesioni edematose e infiltrate, molto fotosensibili, ma prive di tendenza alla cicatrizzazionee con rischio virtualmente nullo di progressione sistemica.
Tra le forme rare si annoverano il lupus verrucoso, il lupus bolloso e il lupus lineare di Blaschko, ognuna con peculiarità cliniche specifiche.
Dermatite Erpetiforme di Duhring
La Dermatite Erpetiforme di Duhring (Dermatitis Herpetiformis, DH) è una malattia autoimmune cronico–recidivante classificata nel più ampio gruppo delle dermatosi bollose autoimmuni, che comprende: pemfigo volgare, pemfigo foliaceo, pemfigoide bolloso, pemfigoide delle mucose, l’epidermolisi bollosa acquisita ed altre. Tali patologie sono tutte accomunate dalla presenza di autoanticorpi diretti contro proteine strutturali della cute.
Nella DH il bersaglio immunologico principale è rappresentato dalla transglutaminasi epidermica (TG3) e, in misura variabile, dalla transglutaminasi tissutale (TG2). Nonostante almeno il 75–90% dei pazienti presenti un’enteropatia glutine‑sensibile compatibile con celiachia, il quadro gastrointestinale è spesso silente; di conseguenza la malattia può manifestarsi inizialmente solo con segni cutanei.
L’età di esordio tipica è tra la seconda e la quarta decade di vita e la distribuzione in base al sesso è sostanzialmente 1:1.
Da un punto di vista fisiopatologico, la frazione gliadinica stimola la mucosa intestinale in soggetti geneticamente suscettibili (HLA‑DQ2/DQ8) inducendo produzione di autoanticorpi di tipo IgA. Gli immuno-complessi IgA anti‑TG3 formano dei depositi granulari nelle papille dermiche e nella giunzione dermo‑epidermica, attivando complemento e reclutamento neutrofilico; l’attività proteasica che ne consegue porta alla degradazione della matrice e allo split subepidermico con formazione delle vescicole tipiche.
Clinica
Comparsa di lesioni papulo‑vescicolari simmetriche intensamente pruriginose. Il decorso della DH è tipicamente cronico con fasi di remissione e di riacutizzazione. Il sintomo cardine è il prurito molto intenso, talvolta urente o doloroso, che precede o accompagna la comparsa delle lesioni.
Le manifestazioni elementari comprendono: papule eritemato-edematose; vescicole e vescico-bolle raggruppate (raramente integre per rottura indotta dal grattamento); erosioni croste ed escoriazioni secondarie al grattamento.
Sedi classiche
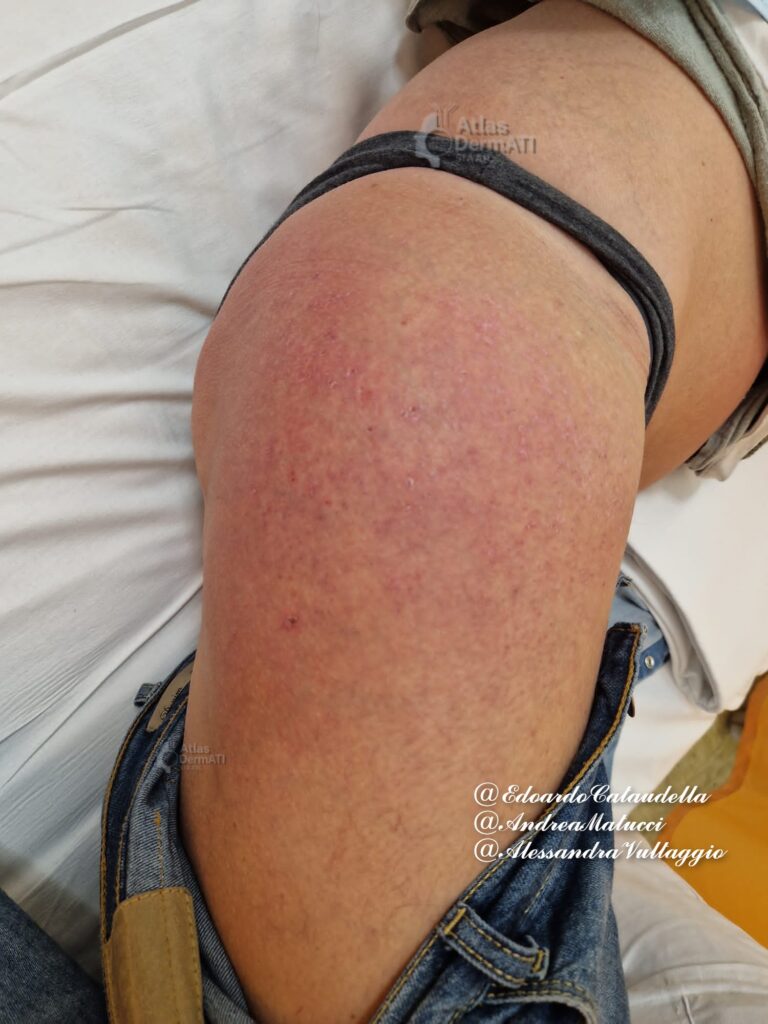
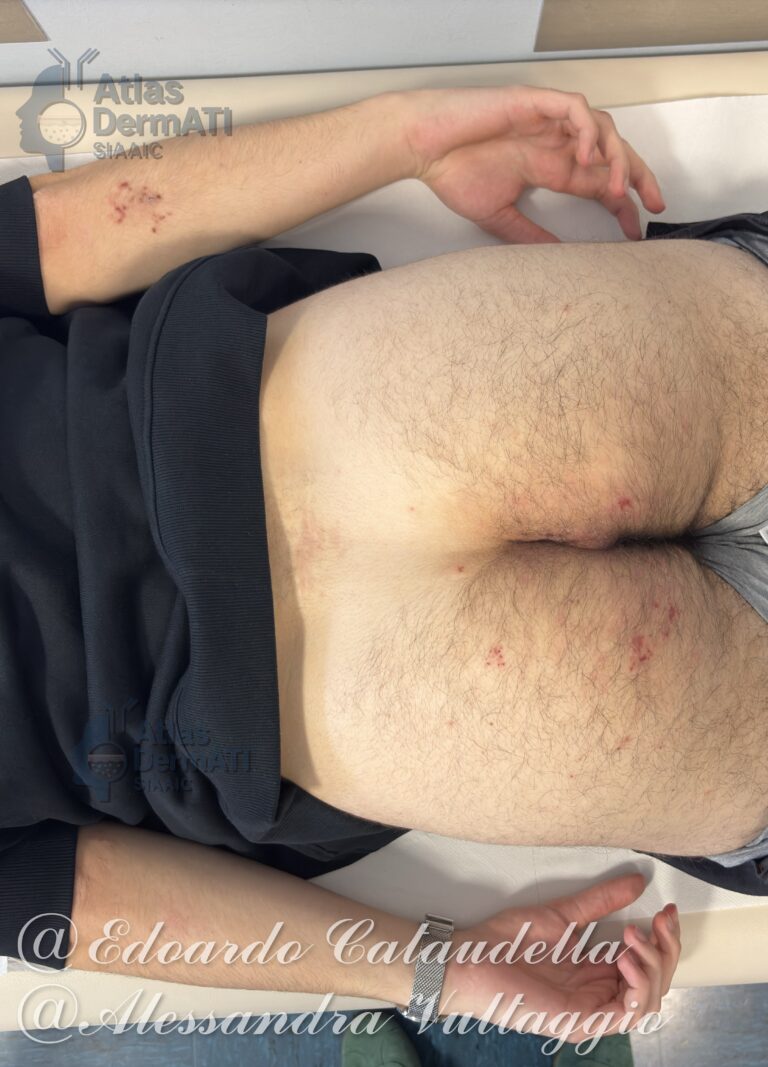
- Superfici estensorie dei gomiti e avambracci;
- Ginocchia;
- Regione glutea;
- Dorso superiore e spalle.
Diagnosi
I test sierologici ELISA per IgA anti‑TG2 e soprattutto anti‑TG3 hanno elevata sensibilità e specificità. Istologicamente, alla colorazione con ematossilina‑eosina, il reperto tipico è costituito da vescicole subepidermiche con microascessi di neutrofili e radi eosinofili nelle papille dermiche. L’IFD rappresenta il test gold standard e mostra depositi granulari di IgA nelle papille e/o lungo la giunzione dermo‑epidermica.
Diagnosi differenziale
La DH può mimare numerose dermatosi più frequenti:
- eczema nummulare o atopico;
- orticaria papulare;
- scabbia;
- pemfigoide bolloso;
- dermatite lineare da IgA;
- lupus eritematoso bolloso;
- psoriasi pustolosa;
Elementi di allarme per altra diagnosi sono: assenza di prurito, burrows interdigitali, lesioni mucose cicatriziali, bolle di grandi dimensioni tese.
Terapia
- Dieta priva di glutine rigorosa indipendentemente dalla coesistenza di interessamento gastrointestinale.
- In caso di lesioni estese e prurito invalidante è indicata la terapia con Dapsone a dosaggio personalizzato, corticosteroidi topici e antistaminici come adiuvanti per un breve periodo di tempo.
Dermatomiosite
La Dermatomiosite (DM) è una malattia autoimmune che si inserisce nel più ampio gruppo delle miopatie infiammatorie idiopatiche (IIMs).
Nella DM il principale bersaglio della malattia è la cute. Tuttavia, quasi sempre, l’interessamento cutaneo è solo l’epifenomeno di una malattia infiammatoria sistemica che può interessare i polmoni, i muscoli e le articolazioni e che nel 6,7-32% dei casi, origina da un fenomeno paraneoplastico.
Esordio generalmente in età adulta (50-60 anni di età) e maggiore frequenza nel sesso femminile (associata ad adenocarcinomi della mammella e dell’ovaio nelle donne e al tumore del polmone sia nelle donne che negli uomini).
Lo spettro delle lesioni cutanee della DM è molto eterogeneo:
- Lesioni Patognomoniche: Papule di Gottron, Segno di Gottron
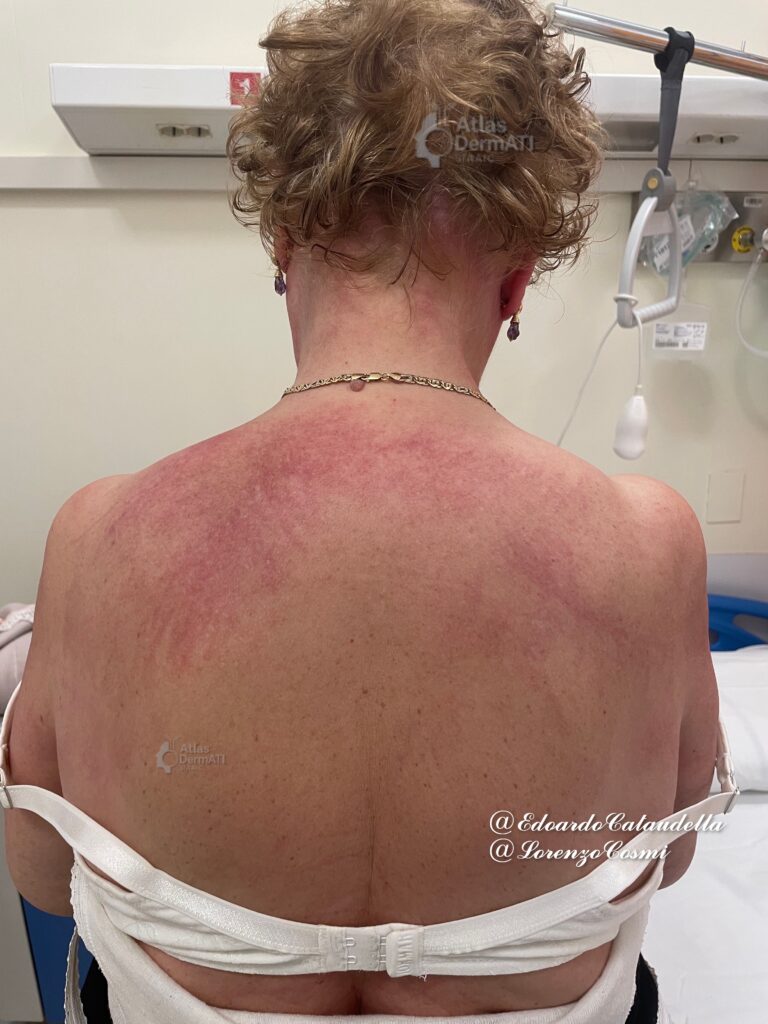
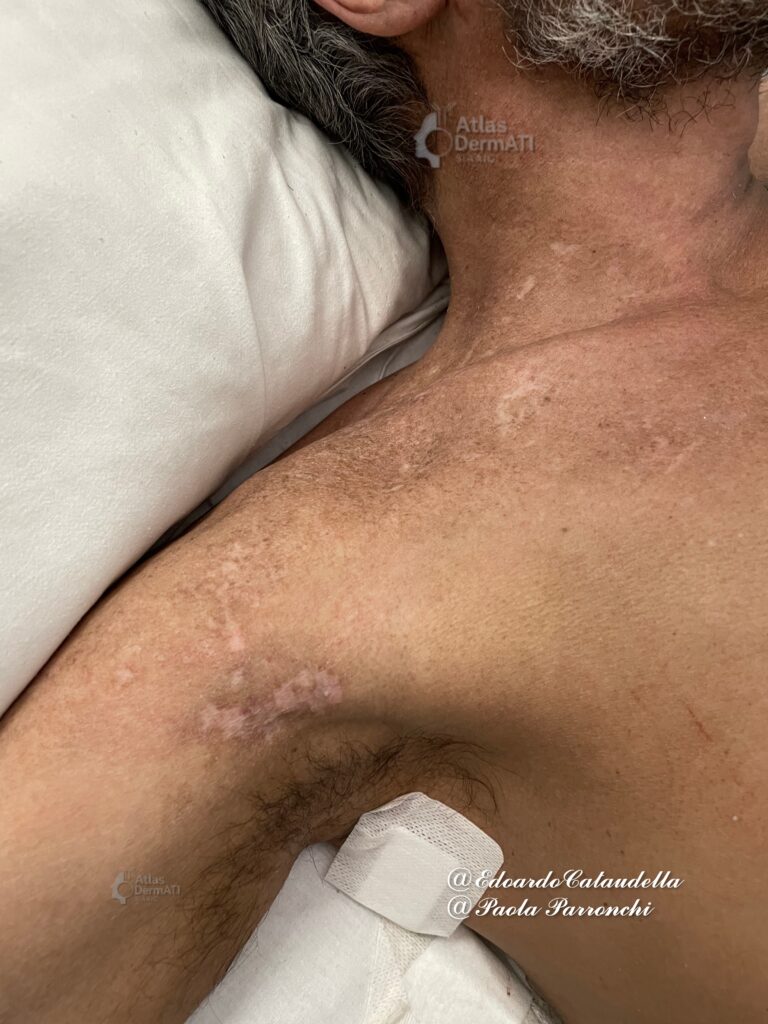
- Lesioni Caratteristiche: Rash eliotropo e edema periorbitale; Telangectasie e eritema periungueali associate a distrofia cuticolare, Eritema cremisi maculare delle superfici estensorie delle mani, dei gomiti, degli avambracci, del torace (Segno del V) delle scapole (segno di Shawl) , delle cosce (Segno di Holster o della fondina) e della fronte.
- Lesioni Compatibili: Poikilodermia vascolare atrofizzante.
- Meno comuni e Rare: Papule di Gottron Inverse, Lesioni ulcerativo necrotiche, vasculite cutanea, calcinosi cutanea, mani da meccanico e piedi da scalatore, eritema flagellato, panniculite, eritrodermia
Le lesioni possono essere pruriginose o dolenti. Le calcificazioni sono più frequenti nella DM giovanile e si riscontrano sulle superfici estensorie a livello di gomiti, regione iliaca e trocanterica.
La DM primitiva o idiopatica ha una prognosi generalmente buona se adeguatamente trattata, fanno eccezione le forme con interessamento vasculitico, le forme con flogosi sistemica aggressiva MAS-Like e quelle con interessamento polmonare a tipo interstiziopatia rapidamente progressiva (RP-ILD).
Le forme di DM secondarie a neoplasia hanno andamento variabile in base allo stadio della neoplasia al momento della diagnosi.
Sedi
Le lesioni cutanee caratteristiche sono: volto, superfici estensorie degli arti superiori, torace superiore, dorso superiore.
Meno frequenti sono le superfici estensorie degli arti inferiori, la regione glutea e il cuoio capelluto.
Diagnosi differenziale
Le manifestazioni cutanee della DM possono mimare altre dermatosi:
- Lupus Eritematoso Sistemico
- Dermatite seborroica
- Rosacea
- Eruzione polimorfa alla luce solare
- Dermatite allergica da contatto (DAC)
- Psoriasi
- Lichen Planus
- Reazioni da farmaco lichenoidi
- Linfoma cutaneo a cellule T
Terapia
La terapia delle manifestazioni cutanee della DM prevede un trattamento immunosoppressivo sistemico:
- Corticosteroidi in bolo
- Corticosteroidi per os a dosaggio di 1mg/kg e lento decalage
- Immunomodulanti: Idrossiclorochina 5 mg/kg
- Immunosoppressori classici (cDMARDs): Ciclosporina 3-5 mg/kg, Metotrexate 0,1-0,3 mg/Kg
- Immunosoppressori biologici (bDMARDs): Rituximab;
- Terapie future: Anifrolumab.
Vitiligine
La vitiligine è il più comune tra i disordini della pigmentazione cutanea con una prevalenza compresa tra 0,5-2% della popolazione mondiale.
E’ dovuta alla distruzione selettiva, immunomediata dei melanociti che si verifica in soggetti predisposti. La disfunzione del sistema anti-ossidante mitocondriale determina un danno cellulare mediato da specie reattive dell’ossigeno ROS che porta alla perdita della tolleranza contro antigeni melanocitari self. La risposta Th17 diretta contro tali antigeni determina un’attivazione citotossica dei linfociti T CD8+ contro i melanociti nonché la produzione di grandi quantità di IFN-gamma anch’esso dannoso per le cellule.
Clinicamente è caratterizzata dalla comparsa di macule ipopigmentate o depigmentate, non desquamanti, color bianco latte a margini netti generalmente asintomatiche e con distribuzione tipica a seconda delle varianti.
Nelle forme non segmentali (NSV) l’esordio è spesso bilaterale e simmetrico, con predilezione per aree soggette a stress meccanico o trauma ripetuto (fenomeno di Koebner).
Decorso
Il decorso è tipicamente cronico, con andamento imprevedibile:
- le chiazze possono rimanere stabili per anni,
- estendersi progressivamente,
- o, meno frequentemente, mostrare re-pigmentazione spontanea.
Nelle forme segmentali (SV) le lesioni cutanee si associano tipicamente a leucotrichia e esordio rapido, la progressione avviene rapidamente nei primi 6–24 mesi, per poi stabilizzarsi nella maggior parte dei casi.
La NSV è associata a un aumentato rischio di comorbidità autoimmuni, in particolare tiroiditi autoimmuni (prevalenza media 15–16%), alopecia areata e, più raramente, altre malattie autoimmuni sistemiche. L’impatto psicologico è spesso significativo, con frequenti sintomi ansioso-depressivi e riduzione della qualità di vita.
Sedi
Qualsiasi area cutanea può essere coinvolta. Le sedi più tipiche includono:
- volto, in particolare regioni periorificiali,
- mani e dita, con interessamento frequente dei polpastrelli,
- gomiti, ginocchia, caviglie,
- regione genitale e mucose,
- cuoio capelluto, talvolta conpoliosi (leucotrichia), segno prognostico sfavorevole per la repigmentazione .
Le forme NSV comprendono varianti cliniche:
- Generalizzata,
- Acrofaciale,
- Mucosale,
- Universale(oltre l’80–90% della superficie corporea BSA),
- Focale
- Varianti rare(follicolare, ipocromica, punteggiata).
La vitiligine segmentale coinvolge tipicamente un singolo dermatomero, spesso a livello del territorio trigeminale sul volto o dei segmenti toracici e degli arti, con pattern talvolta sovrapposti alle linee di Blaschko.
Diagnosi differenziale
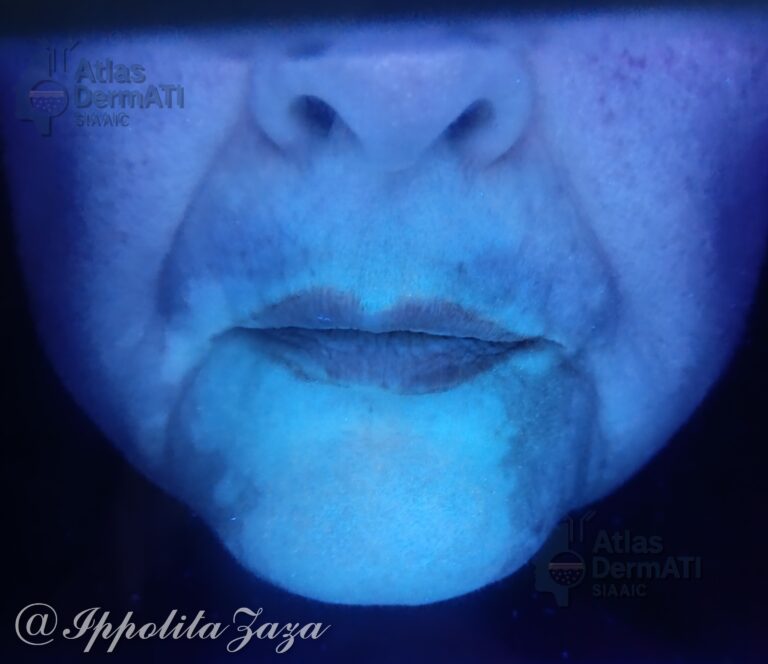
La diagnosi è clinica, supportata dalla lampada di Wood, che evidenzia una fluorescenza bianco-lattea più estesa delle lesioni visibili a occhio nudo, utile anche per l’identificazione dell’attività di malattia. La biopsia è raramente necessaria.
Le principali diagnosi differenziali includono:
- Infettive: Pitiriasi versicolor(fluorescenza giallo-verdastra, esame micologico positivo),
- Infiammatorie: Pitiriasi alba(esordio pediatrico, lieve desquamazione), Leucodermia post-infiammatoria,
- Ipopigmentazione post-traumatica
- Idiopatiche: Ipomelanosi guttata idiopatica, Ipomelanosi maculare progressiva acquisita (fluorescenza follicolare arancio-rossastra alla Wood),
- Congenite: Nevus depigmentosuse altre ipomelanosi congenite.
Marker clinici di attività includono Koebner, lesioni confetti-like, margini eritematosi (vitiligine infiammatoria) e leucotrichia.
Data la frequenza di comorbidità tiroidee, è raccomandato uno screening iniziale e annuale con dosaggio di TSH, anti-TPO e anti-TG.
Terapia
La strategia terapeutica dipende da estensione, attività e forma clinica (NSV vs SV).
Le principali opzioni includono:
Terapie topiche
- Corticosteroidi topici(classe III–IV): prima scelta nelle forme limitate (<3% BSA). Efficaci soprattutto su volto e collo; da usare con cautela per il rischio di atrofia cutanea.
- Inibitori della calcineurina topici(tacrolimus, pimecrolimus): efficaci per volto e collo, sicuri per l’uso prolungato e utili anche come terapia proattiva per prevenire le recidive.
- JAK-i Topici: Ruxolitinib (inibitore di JAK1-2) per uso topico in crema concentrato all’1,5% è stato recentemente introdotto con buoni dati di efficacia e sicurezza soprattutto nel trattamento delle forme limitate a volto e mani.
Fototerapia
- NB-UVB: gold standard nelle forme generalizzate. Percentuali di repigmentazione ≥75% nel 20–35% dei pazienti dopo 6–12 mesi, con massima risposta al volto.
- Laser 308 nm (excimer): utile nelle forme localizzate o segmentali.
Vantaggi: La fototerapia può arrestare la progressione della malattia nelle fasi attive.
Limiti: Efficacia ridotta su mani e piedi.
Terapie sistemiche
- Corticosteroidi sistemici in mini-boliper le forme rapidamente progressive, spesso in combinazione con NB-UVB.
- IJAK-inibitori sistemici emergono come opzione promettente, in quanto colpiscono vie centrali (IFN-γ, JAK/STAT, CXCL10) documentate nella patogenesi della malattia, anche se le indicazioni terapeutiche dipendono dalle approvazioni nazionali.
Terapie chirurgiche
Indicate solo in forme stabili, soprattutto segmentali:
- trapianto di melanociti,
- innesti epidermici a spessore parziale.
Supporto al paziente
- fotoprotezione rigorosa,
- camouflaging cosmetico,
- counseling psicologico e supporto sociale (data l’elevata prevalenza di depressione e ansia).
Sindrome da anticorpi antisintetasi
La sindrome da anticorpi antisintetasi (Anti-Synthetase Syndrome, ASSD) è una malattia autoimmune sistemica rara, appartenente allo spettro delle miopatie infiammatorie idiopatiche (IIMs), caratterizzata dalla presenza di autoanticorpi diretti contro le aminoacil-tRNA sintetasi.
La malattia presenta un’elevata eterogeneità clinica e un decorso variabile, con coinvolgimento multisistemico.
Classicamente caratterizzata da una triade clinica (raramente presente al completo all’esordio della malattia): interessamento polmonare interstiziale (ILD), miopatia infiammatoria e artrite. Oltre alla triade, sono comuni febbre di origine indeterminata, fenomeno di Raynaud e manifestazioni cutanee.
Età di esordio: generalmente adulta, con una lieve prevalenza nel sesso femminile. L’andamento clinico è fortemente condizionato dalla presenza e dalla gravità dell’interstiziopatia polmonare, che rappresenta il principale determinante prognostico.
Interessamento cutaneo
Le manifestazioni cutanee dell’ASSD rivestono un ruolo clinico rilevante, sia come segno suggestivo di malattia sia per il loro valore nel differenziare l’ASSD da altre IIM, in particolare la dermatomiosite.
L’interessamento cutaneo può precedere, accompagnare o seguire le manifestazioni sistemiche ed è presente in una quota significativa di pazienti.
Lesioni caratteristiche e patognomoniche
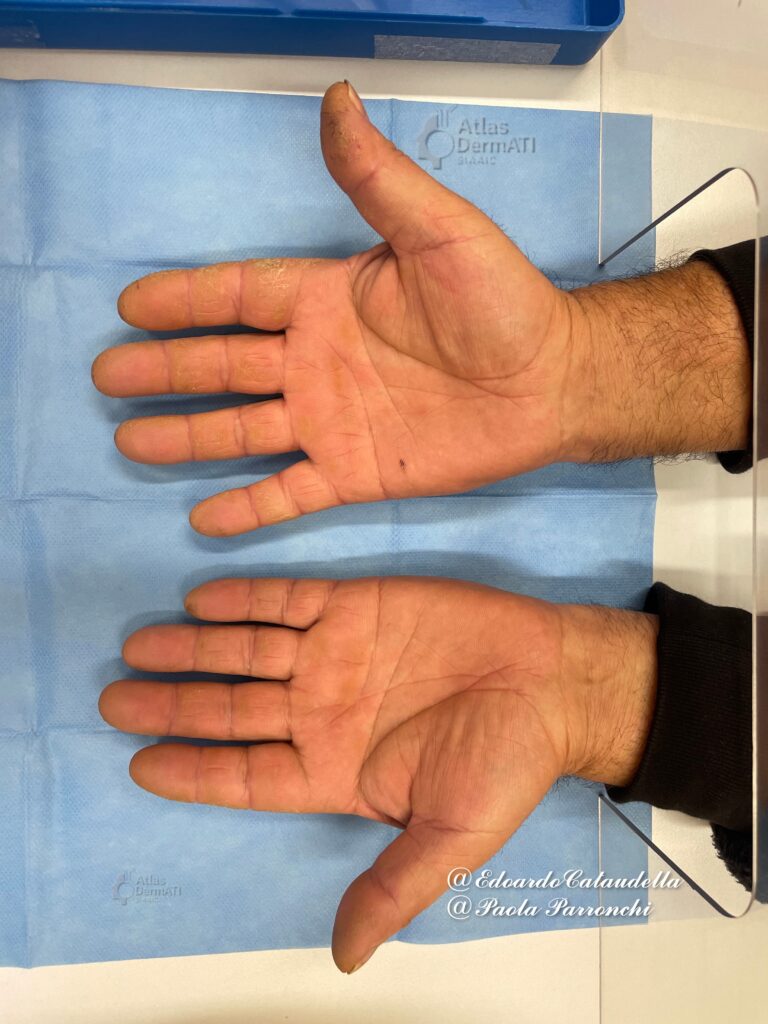
La manifestazione cutanea più tipica dell’ASSD è rappresentata dalle cosiddette “mani da meccanico” (mechanic’s hands). Si tratta di lesioni ipercheratosiche, fissurate, eritematose, prevalentemente localizzate sui margini laterali delle dita e del pollice, con relativo risparmio delle superfici palmari e dorsali. Le lesioni tendono a fluttuare parallelamente all’attività di malattia.
Una manifestazione analoga, a carico dei piedi, è rappresentata dagli “hiker’s feet” (piedi da escursionista).
Lesioni di tipo dermatomiositico
Circa il 20% dei pazienti con ASSD presenta manifestazioni cutanee sovrapponibili a quelle della dermatomiosite: papule e segno di Gottron; rash eliotropo; eritema delle superfici estensorie (segno dello Shawl, segno del V, segno della fondina); alterazioni periungueali con eritema e teleangectasie; ipertrofia e distrofia delle cuticole.
Altre manifestazioni cutanee
- Fenomeno di Raynaud;
- Rash psoriasiformi o eczematosi;
- livedo, porpora o altre manifestazioni vascolari cutanee, più rare.
Sedi di interessamento cutaneo
- mani (margini laterali delle dita) e, meno frequentemente, piedi;
- superfici estensorie degli arti superiori;
- volto e regione perioculare (nei casi con rash tipo eliotropo);
- torace superiore e dorso superiore nelle forme con Dermatite di Gottron.
Diagnosi differenziale
- Dermatomiosite;
- Lupus eritematoso sistemico e cutaneo;
- Psoriasi e dermatiti eczematose croniche;
- Dermatite da contatto allergica o irritativa (DAC e DIC) delle mani;
- Sclerosi sistemica (in presenza di Raynaud e alterazioni periungueali).
La ricerca degli autoanticorpi antisintetasi, unitamente alla valutazione del coinvolgimento polmonare, muscolare e articolare, è fondamentale per la corretta classificazione.
Terapia
Il trattamento dell’interessamento cutaneo nell’ASSD si inserisce nella gestione globale della malattia sistemica e si basa prevalentemente su terapie immunosoppressive.
Le opzioni terapeutiche includono:
- corticosteroidi sistemici, spesso utilizzati come terapia di induzione;
- immunosoppressori convenzionali (cDMARDs) quali metotrexate, azatioprina, micofenolato mofetile o ciclosporina;
- immunosoppressori biologici (bDMARDs), in particolare rituximab, nei casi refrattari;
- farmaci antimalarici (es. idrossiclorochina) come terapia di supporto per il controllo delle manifestazioni cutanee, soprattutto nelle forme con rash di tipo dermatomiositico.
Le terapie topiche (corticosteroidi o immunomodulatori topici) possono essere utilizzate come trattamento adiuvante per le lesioni cutanee localizzate, ma raramente risultano sufficienti come monoterapia.
Pemfigo Volgare
Il pemfigo volgare è una malattia autoimmune bollosa intraepidermica a decorso cronico e recidivante, potenzialmente letale. È causato dalla produzione di autoanticorpi IgG diretti contro le desmogleine, proteine fondamentali per l’adesione tra i cheratinociti; la loro alterazione determina acantolisi e conseguente formazione di bolle.
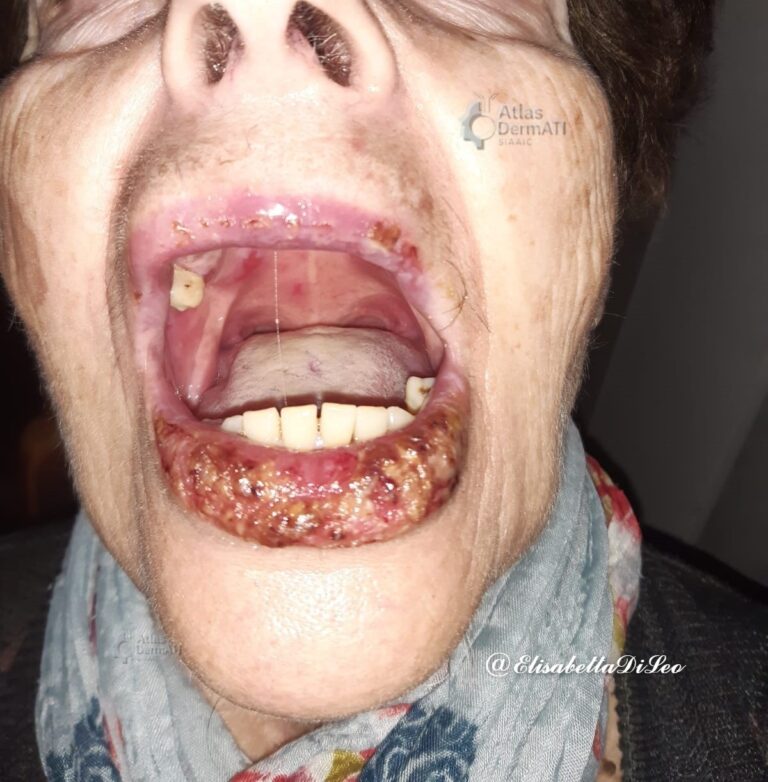
La lesione elementare del pemfigo è rappresentata da una bolla che insorge su cute aflegmasica, cioè non eritematosa, a cupola flaccida, piene di fluido chiaro. Le bolle, a causa della fragilità del tetto, vanno frequentemente incontro a rottura e residuano aree esulcerate circondate da un collaretto epiteliale. Il dolore, soprattutto a livello orale, può compromettere l’alimentazione e le attività quotidiane, con importanti ripercussioni sulla qualità di vita e sul benessere psicologico del paziente.
Sedi
Nel pemfigo volgare il coinvolgimento mucosale è estremamente frequente e rappresenta nella maggior parte dei casi la prima manifestazione clinica della malattia. Circa il 90% dei pazienti presenta lesioni della mucosa orale.
Sedi più comunemente interessate: mucosa buccale e labiale, meno frequentemente coinvolte le mucose di orofaringe, esofago, congiuntiva e vulva.
Il coinvolgimento cutaneo è caratterizzato dalla comparsa di bolle su cute normale o eritematosa. Le lesioni cutanee possono essere localizzate o generalizzate, con una predilezione per scalpo, volto, tronco, ascelle e regione inguinale; palmi e piante sono generalmente risparmiati. In caso di interessamento dello scalpo può comparire alopecia. Nella maggior parte dei pazienti è presente il segno di Nikolsky positivo, espressione della fragilità epidermica. Le erosioni cutanee tendono a guarire senza esiti cicatriziali.
Diagnosi
La diagnosi di pemfigo volgare è confermata dalla biopsia di aree di pelle lesionata e circostante normale (perilesionale). All’esame istologico si osserva una bolla intraepidermica sovrabasale, con acantolisi e distacco dei cheratinociti dagli strati sottostanti. Il test di immunofluorescenza diretta mostra autoanticorpi IgG contro la superficie cellulare dei cheratinociti.
Autoanticorpi sierici verso le glicoproteine transmembrana, desmogleina 1 e desmogleina 3, possono essere identificati mediante immunofluorescenza diretta, immunofluorescenza indiretta e il test ELISA. La sensibilità del test ELISA è più alta (> 95% di quella dell’immunofluorescenza indiretta). Il dosaggio degli autoanticorpi è utile anche per la valutazione dell’attività di malattia e del follow-up terapeutico.
Diagnosi differenziale
La diagnosi differenziale del pemfigo volgare comprende altre dermatosi bollose e patologie ulcerative muco-cutanee:
- pemfigoide bolloso, che si distingue per la presenza di bolle a tetto teso, prurito intenso, segno di Nikolsky negativo e depositi lineari di IgG e C3 lungo la giunzione dermo-epidermica;
- stomatite aftosa;
- eritema multiforme;
- lichen planus;
- pemfigo paraneoplastico;
- pemfigo foliaceo, quest’ultimo caratterizzato dall’assenza di coinvolgimento mucoso.
La distinzione si basa sull’aspetto clinico, sull’andamento della malattia e soprattutto sui reperti immunopatologici.
Terapia
La terapia del pemfigo volgare ha come obiettivo l’induzione della remissione clinica, limitando al contempo il rischio di complicanze legate sia alla malattia sia al trattamento.
Terapia di prima linea: corticosteroidi sistemici (es: prednisone, somministrato inizialmente a dosi elevate e successivamente ridotto in modo graduale in base alla risposta clinica).
Al fine di limitare gli effetti collaterali degli steroidi, vengono frequentemente associati: immunosoppressori adiuvanti come azatioprina o micofenolato mofetile.
Il rituximab, anticorpo monoclonale anti-CD20, ha rivoluzionato la gestione della malattia, consentendo elevate percentuali di remissione completa e la riduzione o sospensione dei corticosteroidi.
Nei casi refrattari possono essere utilizzate immunoglobuline endovena o altre terapie di terza linea. Fondamentale è anche la terapia di supporto, con cura delle lesioni, prevenzione delle infezioni, controllo del dolore e supporto nutrizionale.