Miscellanea di dermatosi di interesse immuno-allergologico
Coordinamento a cura di Alessandro Sinisi e Remo Poto
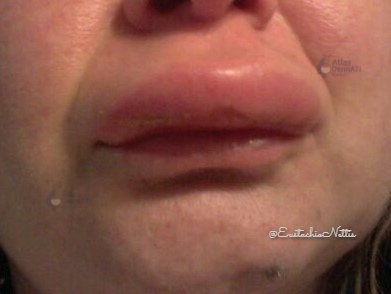
Cheilite granulomatosa
La cheilite granulomatosa è una condizione infiammatoria cronica rara, inquadrata nello spettro delle orofacial granulomatoses (OFG), caratterizzata clinicamente da tumefazione labiale ricorrente o persistente, in genere paucisintomatica o non dolente, e istologicamente da infiammazione granulomatosa non caseosa quando presente. È considerata una variante dell’OFG e può rappresentare una forma incompleta della sindrome di Melkersson-Rosenthal (MRS), nella quale l’edema orofacciale può associarsi a paralisi del nervo facciale e lingua plicata (o lingua scrotale).
Sintomatologia
E’ dominata dall’edema labiale, spesso con esordio acuto e andamento inizialmente intermittente, che tende successivamente a diventare persistente e progressivo, con indurimento e deformità funzionali ed estetiche. Quando la tumefazione è marcata, possono comparire fissurazioni mediane e ai commessurali (cheilite angolare) e, in una quota di pazienti, una sensazione urente; nei quadri OFG più severi possono associarsi ulcerazioni orali e linfoadenopatia latero-cervicale.
Sedi
La localizzazione più frequente è rappresentata da uno o entrambi i labbri. Meno frequentemente: nell’OFG possono essere coinvolte la mucosa delle guance, le gengive, il palato, il pavimento orale e la lingua, nonché aree periorali, zigomatiche e frontali.
Diagnosi
E’ necessaria la correlazione clinico-istologica e un iter di esclusione. Istologicamente, il tessuto può mostrare edema subepiteliale, dilatazione linfatica, infiltrato granulomatoso e granulomi non necrotici con componente linfocitaria ed elementi epitelioidi, talora con cellule giganti e fibrosi; tuttavia, la biopsia può evidenziare anche infiammazione cronica aspecifica e l’assenza di granulomi non deve escludere automaticamente la diagnosi, soprattutto nelle fasi iniziali o nei campioni non rappresentativi..
Diagnosi differenziale
Utile considerare nella diagnostica differenziale condizioni di tumefazione labiale transitoria, quali angioedema (anche non istamino mediato), traumi e infezioni comuni, ma soprattutto entità che mimano un edema persistente o recidivante. In particolare, è necessario escludere malattie granulomatose sistemiche e condizioni infettive granulomatose: nella pratica clinica vanno considerate e investigate almeno malattia di Crohn, sarcoidosi, tubercolosi e, in contesti epidemiologici appropriati, leishmaniosi e lebbra.
Terapia e trattamenti
- i corticosteroidi costituiscono il cardine terapeutico: la somministrazione intralesionale è frequentemente impiegata per ottenere elevate concentrazioni locali e può determinare risposte buone o parziali, spesso con necessità di cicli ripetuti;
- gli steroidi sistemici possono migliorare alcune manifestazioni (in particolare ulcerazioni) ma meno costantemente la macrocheilia. Le recidive alla sospensione sono comuni;
- segnalato l’impiego di antibiotici con proprietà immunomodulanti (macrolidi, tetracicline, metronidazolo), farmaci immunomodulanti e immunosoppressori (ad esempio metotrexate o azatioprina in contesti selezionati, specie se associati a malattia di Crohn);
- nei casi refrattari l’impiego di farmaci biologici, in particolare anti-TNFα (infliximab, adalimumab), ha mostrato risposte variabili;
- nei pazienti con macrocheilia recalcitrante e importante compromissione funzionale, la cheiloplastica riduttiva rappresenta un’opzione efficace, talora con risultati persistenti.
Pioderma Gangrenoso
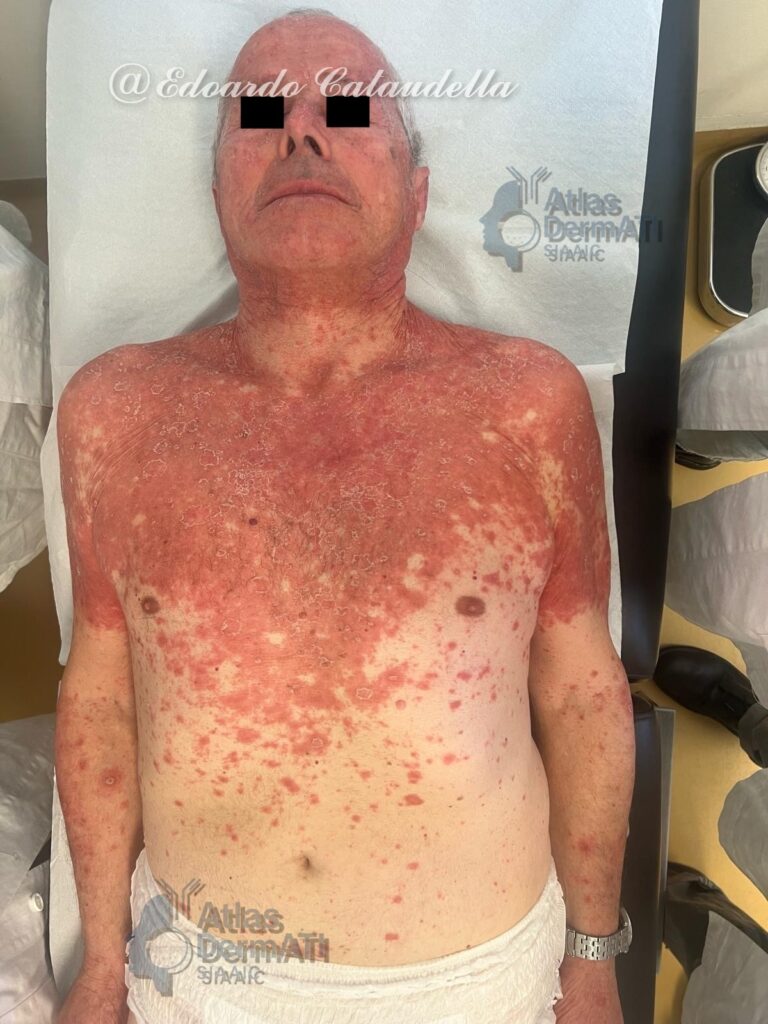
Il pioderma gangrenoso (PG) è una rara dermatosi infiammatoria non infettiva appartenente al gruppo delle dermatosi neutrofiliche. Può presentarsi come forma idiopatica oppure associarsi a patologie sistemiche, in particolare malattie infiammatorie croniche intestinali, artriti infiammatorie e disordini ematologici.
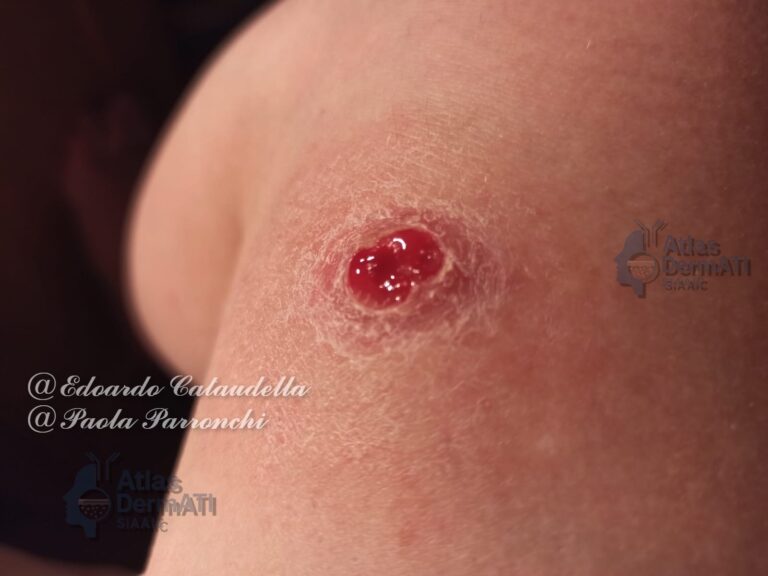
Clinica: ulcerazioni cutanee dolorose a rapida evoluzione, con margini irregolari, sottominati e di colore eritemato-violaceo.
Patogenesi complessa e multifattoriale, basata su una profonda disregolazione dell’immunità innata e adattativa, con predominanza di una risposta Th1/Th17 e iperattivazione neutrofilica, spesso in soggetti geneticamente predisposti all’autoinfiammazione.
Un elemento caratteristico della malattia è il fenomeno della patergia, per cui traumi cutanei sono i principali trigger. La diagnosi è prevalentemente clinica e rimane complessa per l’assenza di reperti istopatologici patognomonici e per l’ampio spettro di condizioni che possono mimarne la presentazione. Anche l’approccio terapeutico attuale è scarsamente strutturato.
Sintomatologia
Spesso esordio rapido cui può seguire una fase di spegnimento o una fase di cronicizzazione ad andamento relapsing-remitting soprattutto in presenza di patologie sistemiche associate. Le lesioni precoci possono presentarsi come papule, pustole o noduli infiammatori dolorosi, che evolvono rapidamente in ulcerazioni necrotiche a margini irregolari, sottominati e di colore eritemato-violaceo, con fondo fibrino-necrotico. Il dolore è generalmente intenso e sproporzionato rispetto all’estensione clinica della lesione. La guarigione della singola ulcera avviene lentamente e può lasciare esiti cicatriziali cribrosi o atrofici.
Sedi
Il PG può interessare qualsiasi distretto cutaneo, ma colpisce più frequentemente gli arti inferiori, in particolare la superficie anteriore delle gambe. Altre sedi comuni includono il tronco, gli arti superiori e le regioni peristomali. Esistono diverse varianti cliniche, ciascuna con una distribuzione preferenziale: la forma ulcerativa classica predilige gli arti inferiori; la variante bollosa interessa più spesso volto e arti superiori ed è frequentemente associata a patologie ematologiche; la forma pustolosa è tipicamente correlata a malattie infiammatorie intestinali; la variante vegetante presenta un decorso più indolente ed è solitamente limitata al tronco; il PG peristomale si sviluppa in prossimità di stomie chirurgiche. Sono descritte anche forme post-operatorie localizzate alle sedi di incisione chirurgica.
Diagnosi differenziale
La diagnosi differenziale del PG è ampia: ulcere vascolari di origine arteriosa o venosa, vasculiti sistemiche, trombosi cutanee, calcifilassi, infezioni batteriche, micotiche o micobatteriche, nonché neoplasie cutanee primitive o secondarie.
Altre condizioni da considerare includono: ulcere farmacologiche, dermatosi infiammatorie croniche e lesioni factizie.
L’esame istopatologico, pur mostrando un infiltrato neutrofilico dermico, non è specifico e risulta utile principalmente per escludere altre cause. Negli ultimi anni sono stati proposti criteri diagnostici strutturati e score clinici validati, come il PARACELSUS score e i criteri Delphi, che consentono una diagnosi più accurata e riducono il rischio di misdiagnosi, particolarmente frequente nelle ulcere degli arti inferiori.
Terapia e trattamenti
La diagnosi differenziale del PG è ampia: ulcere vascolari di origine arteriosa o venosa, vasculiti sistemiche, trombosi cutanee, calcifilassi, infezioni batteriche, micotiche o micobatteriche, nonché neoplasie cutanee primitive o secondarie.
Altre condizioni da considerare includono: ulcere farmacologiche, dermatosi infiammatorie croniche e lesioni factizie.
L’esame istopatologico, pur mostrando un infiltrato neutrofilico dermico, non è specifico e risulta utile principalmente per escludere altre cause. Negli ultimi anni sono stati proposti criteri diagnostici strutturati e score clinici validati, come il PARACELSUS score e i criteri Delphi, che consentono una diagnosi più accurata e riducono il rischio di misdiagnosi, particolarmente frequente nelle ulcere degli arti inferiori.
Pitiriasi Rubra Pilare
La pitiriasi rubra pilare è una dermatosi infiammatoria cronica rara, caratterizzata da un’alterazione del processo di cheratinizzazione. Si manifesta clinicamente con papule follicolari cheratosiche, chiazze eritemato-desquamative di colorito arancio-rossastro e, nei casi più estesi, con eritrodermia associata a tipiche aree di cute risparmiata.
Sintomatologia e decorso
Presenta un decorso clinico variabile e comprende diverse forme cliniche, distinte in base all’età di insorgenza, all’estensione delle lesioni e all’evoluzione nel tempo. Esistono forme giovanili ereditarie e forme adulte sporadiche, scatenate a volte da infezioni o traumi.
Sedi
Superficie dorsale delle dita delle mani, gomiti, ginocchia, cuoio capelluto, ipercheratosi palmoplantare ed alterazioni ungueali.
Diagnosi
La diagnosi è principalmente clinica, supportata dall’esame istopatologico.
Diagnosi differenziale
Eritrodermia da farmaci, psoriasi eritrodermiforme, eczema severo eritrodermico, dermatite seborroica, ittiosi, linfoma cutaneo a cellule T, lupus cutaneo subacuto.
Terapia
Il trattamento spesso richiede retinoidi orali come l’acitretina, farmaci biologici utilizzati off-label (ustekinumab, etanercept) o immunosoppressori, e mira a gestire i sintomi.
La guarigione completa è rara, specialmente nelle forme adulte.
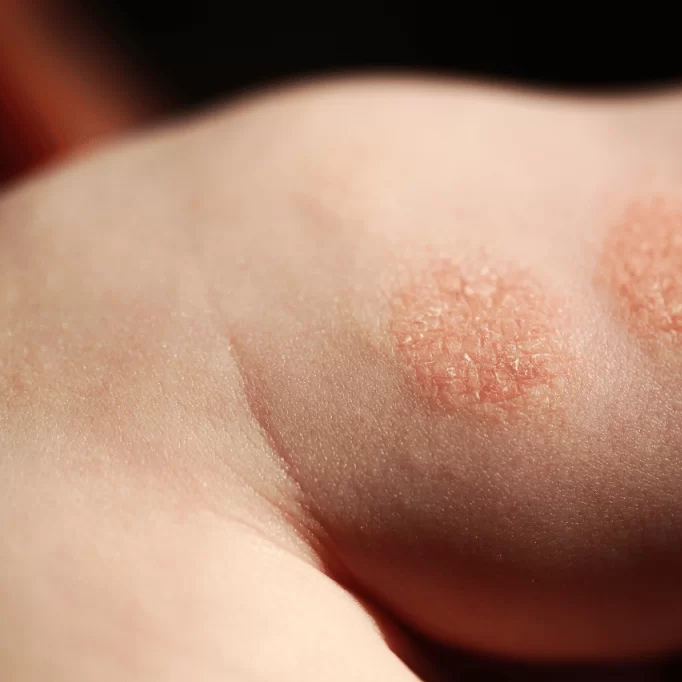
Granuloma anulare
Il granuloma anulare (GA) è una patologia infiammatoria cutanea, benigna e non contagiosa, caratterizzata da papule disposte ad anello attorno ad una zona di cute normale o lievemente depressa. Le lesioni ad anello di colore roseo, rosso o violaceo, si localizzano spesso su mani, piedi, polsi e caviglie.
Tra gli eventi scatenanti segnalati figurano numerose infezioni e infestazioni cutanee e diversi tipi di traumi cutanei. L’infiammazione è mediata dal fattore di necrosi tumorale alfa (TNFα).
Sintomi
Il granuloma anulare può essere asintomatico, ma le aree colpite sono spesso dolenti alla palpazione. Le placche tendono a cambiare lentamente forma, dimensione e posizione.
I segni e i sintomi del granuloma anulare possono variare a seconda della distribuzione e localizzazione delle lesioni:
GA localizzato
Forma più comune con bordi circolari o semicircolari, con un diametro fino a 5 centimetri. Sedi preferenziali: mani (maggiormente dita, nocche e dorso), piedi, polsi e caviglie nei bambini e giovani adulti. Il granuloma anulare generalizzato si manifesta solitamente negli adulti con chiazze diffuse di colore rosato o leggermente malva.
GA generalizzato
È raro, colpisce solitamente gli adulti e può causare prurito. È caratterizzato da piccole papule, solitamente disposte simmetricamente in anelli poco definiti di 10 cm o più di diametro. Le lesioni si presentano spesso in prossimità delle pieghe cutanee del tronco (ascelle, inguine). Questa forma può associarsi ad HIV.
GA sottocutaneo
Presente solitamente nei bambini piccoli. In questo caso, anziché un’eruzione cutanea, il GA è caratterizzato da piccoli noduli sodi sottocutanei che interessano mani, superficie anteriore della gamba e cuoio capelluto.
Cause
Non note. Fattori scatenanti potrebbero essere:
- Punture insetti, infezioni, esposizione al sole, lesioni cutanee minori, farmaci.
Tuttavia, sono state segnalate diverse associazioni con patologie sistemiche, tra cui tiroidite autoimmune, diabete mellito, altre malattie autoimmuni organo-specifiche, iperlipidemia e, raramente, linfoma, infezione da HIV e tumori solidi.
Diagnosi
La diagnosi è solitamente clinica per il suo aspetto caratteristico. Nei casi dubbi la biopsia cutanea mostra solitamente una degenerazione necrobiotica del collagene dermico circondata da una reazione infiammatoria.
Trattamento
Nella maggior parte dei casi, il GA non richiede trattamento poiché le lesioni scompaiono spontaneamente in pochi mesi, senza lasciare esiti. Tuttavia, se non trattato, il GA può durare alcune settimane o anni.
Le opzioni terapeutiche da considerare includono:
- Pomate corticosteroidee topiche con bendaggio occlusivo
- Iniezioni intralesionali di steroidi
- Crioterapia o ablazione laser
- Inibitori topici della calcineurina (tacrolimus e pimecrolimus).
La terapia sistemica può essere presa in considerazione in caso di GA diffuso sebbene nessuno di questi trattamenti può essere considerato efficace per la sua completa risoluzione:
– Steroidi sistemici, Isotretinoina, Metotrexato, Dapsone, Idrossiclorochina, Pentossifillina, Combinazione di antibiotici: rifampicina, ofloxacina, minociclina; Fotochemioterapia (PUVA), Terapia fotodinamica, Ciclosporina, Farmaci biologici, in particolare inibitori del TNFα, come adalimumab e infliximab; inibitori orali e topici della Janus chinasi (JAK), incluso il tofacitinib.
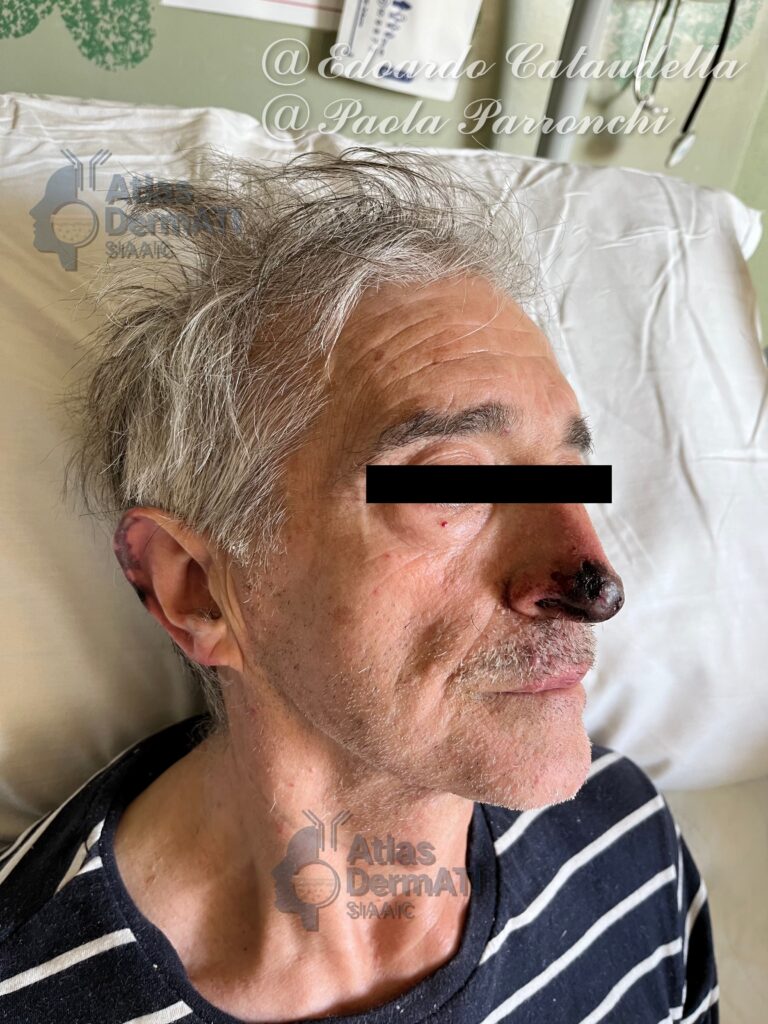
Tofi gottosi
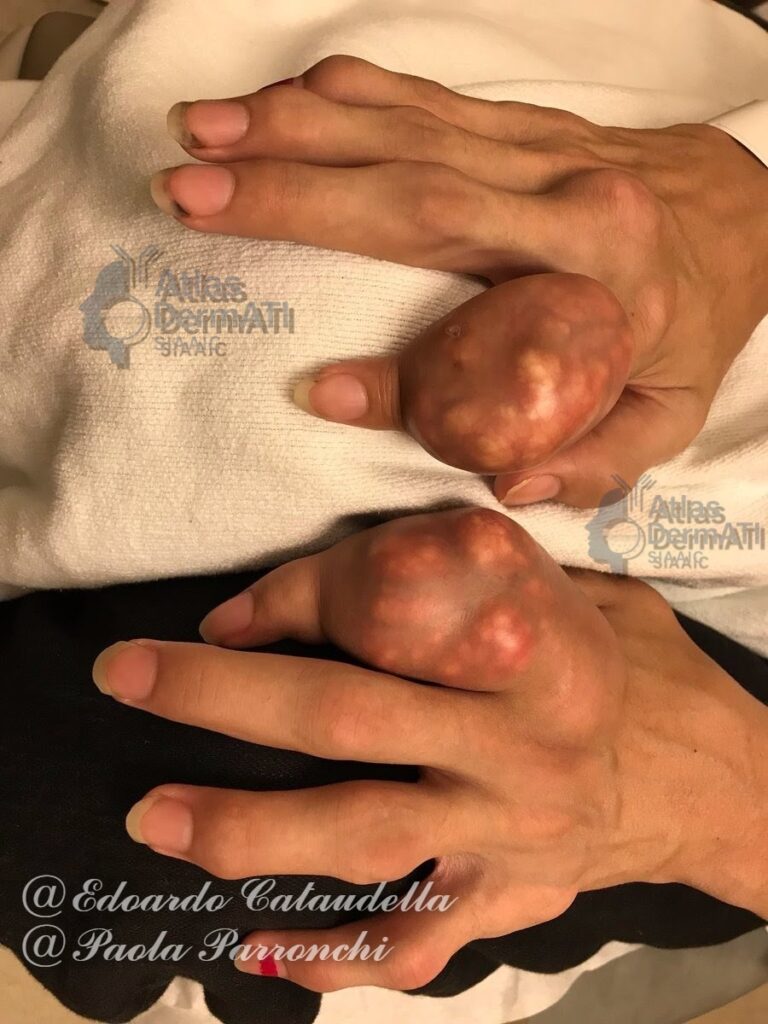
I tofi gottosi rappresentano la manifestazione tipica della gotta cronica, conseguente alla deposizione tissutale di cristalli di urato monosodico in condizioni di iperuricemia persistente. Si configurano come noduli sottocutanei o periarticolari, spesso a crescita lenta, che riflettono una risposta infiammatoria granulomatoide nei confronti dei cristalli e possono associarsi a danno strutturale articolare e dei tessuti molli.
Sintomatologia
I tofi si presentano in genere come lesioni nodulari solide, di consistenza variabile, talora biancastre o giallastre, non necessariamente dolenti. Possono diventare sintomatici per compressione di strutture contigue, limitazione funzionale, dolore meccanico o per la comparsa di complicanze locali. Nei quadri più avanzati possono ulcerarsi, con fuoriuscita di materiale gessoso, e sovrainfettarsi, determinando dolore, essudazione e ritardo di guarigione.
Sedi
La localizzazione più comune interessa regioni periarticolari e aree sottoposte a microtrauma e basse temperature. Tipicamente sono coinvolti padiglioni auricolari, olecrano, dita delle mani e dei piedi, articolazioni metatarso falangee, tendine d’Achille, borse sierose e superfici estensorie. In forme severe possono essere presenti depositi in sedi meno comuni, incluse strutture tendinee profonde e, raramente, organi interni.
Diagnosi
La diagnosi si basa sulla correlazione clinica e, quando necessario, su conferma laboratoristica e strumentale. L’identificazione di cristalli di urato monosodico in aspirato o materiale tofaceo, mediante microscopia a luce polarizzata, costituisce il riferimento diagnostico. L’imaging può supportare l’inquadramento e la valutazione del burden, con ecografia, che può evidenziare depositi e segni suggestivi, e TC dual energy, utile per la caratterizzazione dei depositi di urato e per la quantificazione. La valutazione dell’uricemia è rilevante per la gestione, ma non è sufficiente da sola per porre diagnosi.
Diagnosi differenziale
Include noduli reumatoidi, calcinosi cutanea, xantomi, gangli sinoviali, tumori dei tessuti molli, cisti epidermiche, borsiti croniche e, in presenza di ulcerazione, lesioni infette o croniche di altra natura. In sedi atipiche o in caso di rapida crescita può essere indicato un approfondimento istologico per escludere patologie alternative.
Terapia e trattamenti
L’obiettivo principale è la riduzione stabile dell’uricemia mediante terapia urato abbassante, con progressiva regressione dei tofi e prevenzione delle riacutizzazioni. Il controllo delle fasi infiammatorie acute e delle recidive, inclusa la profilassi nelle fasi di avvio o intensificazione della terapia ipouricemizzante, è parte integrante della gestione. Nei casi con tofi voluminosi, complicati, ulcerati o causa di severa limitazione funzionale, può essere indicata la rimozione chirurgica selettiva, integrata in un percorso di controllo metabolico a lungo termine per prevenire la riformazione dei depositi.
Malattia di Anderson-Fabry
La malattia di Anderson–Fabry è una patologia lisosomiale ereditaria rara, a trasmissione X-linked, causata dal deficit dell’enzima a-galattosidasi A, con conseguente accumulo di globotriaosilceramide (Gb3) nei tessuti.
Sintomatologia e decorso
L’esordio clinico avviene generalmente nell’infanzia o adolescenza con dolore neuropatico acroparestesico, ipoidrosi/anidrosi e intolleranza al calore.
Il decorso è cronico e progressivo, con interessamento multisistemico che coinvolge cute, rene, apparato cardiovascolare e sistema nervoso centrale.
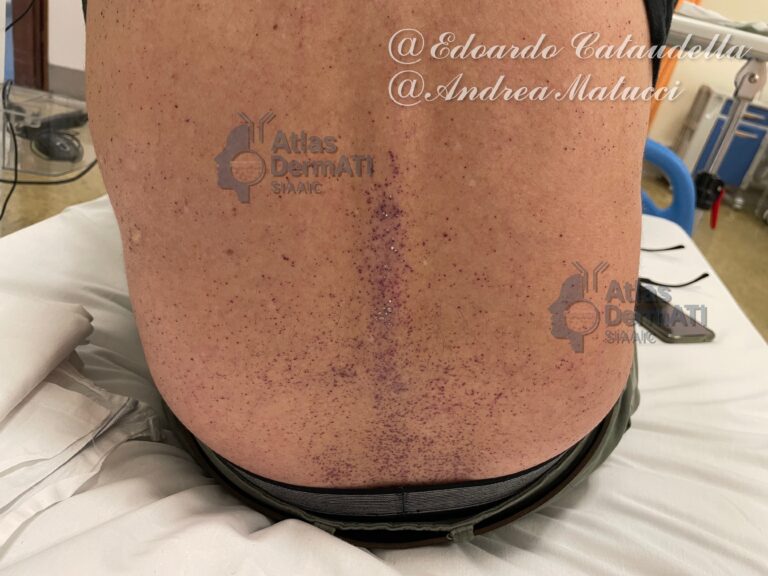
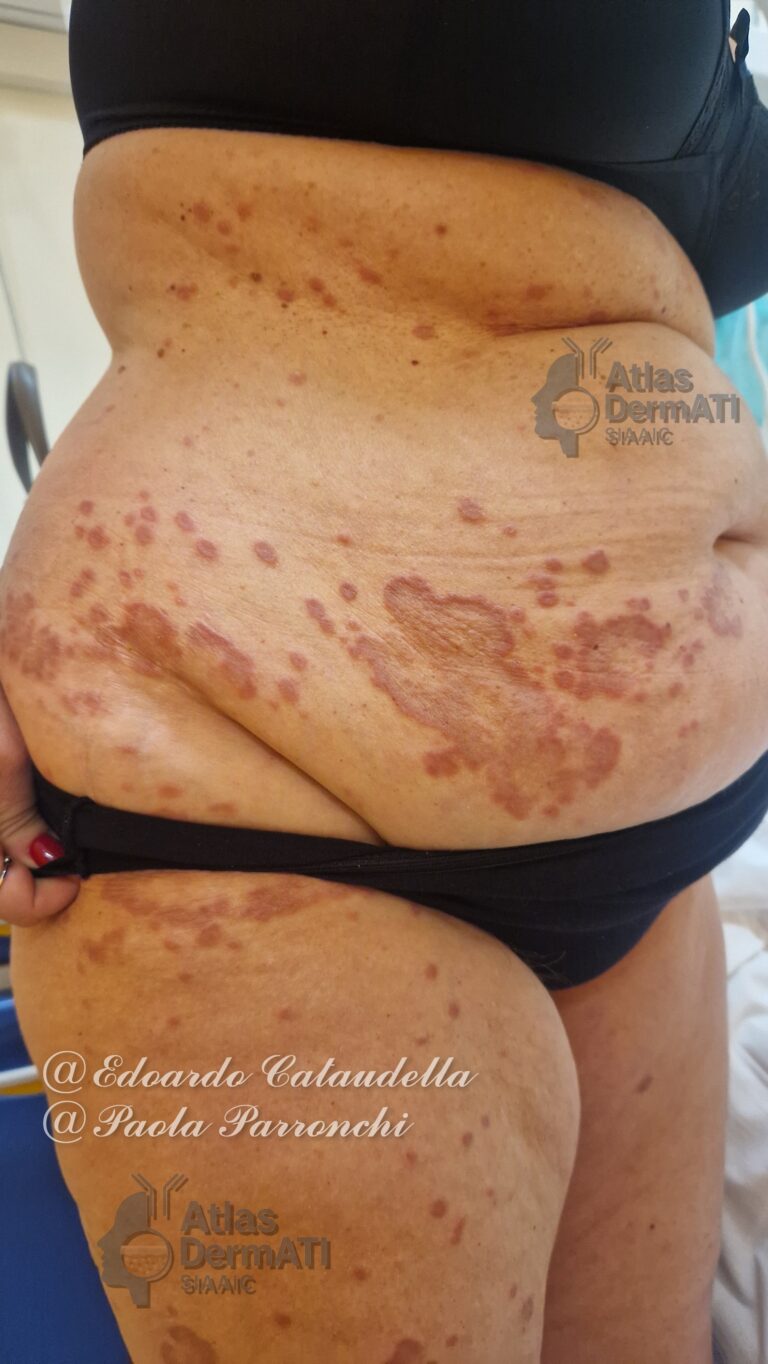
Gli angiocheratomi multipli rappresentano un segno clinico precoce e caratteristico, spesso rilevante per il sospetto diagnostico. Sono lesioni cutanee multiple, puntiformi o confluenti, di colore rosso-violaceo scuro, talora con superficie lievemente ipercheratosica.
Sedi
La distribuzione caratteristica interessa la regione periombelicale, inguinale, glutea e genitale, configurando la tipica “distribuzione a costume da bagno”. Possibile interessamento di mucose e congiuntive.
Diagnosi e accertamenti
La diagnosi è confermata dal dosaggio dell’attività dell’a-galattosidasi A (ridotta o assente nei maschi) e dall’analisi genetica del gene GLA. L’esame istologico cutaneo mostra dilatazioni vascolari del derma superficiale con accumulo lisosomiale nelle cellule endoteliali.
Diagnosi differenziale
La diagnosi differenziale include angiocheratomi isolati (es. angiocheratomi di Fordyce), angiomi capillari, porpora vascolare e altre malattie da accumulo lisosomiale prive di coinvolgimento sistemico.
Terapia
Il trattamento si basa sulla terapia enzimatica sostitutiva con a-galattosidasi A ricombinante e, inpazienti selezionati, sulla terapia chaperonica orale. Le lesioni cutanee non richiedono trattamento specifico, ma possono essere trattate con laser vascolari per indicazioni estetiche o funzionali. La gestione è multidisciplinare e a lungo termine.