Angioedema ereditario
Coordinamento a cura di Erminia Giordano
Sintomatologia e decorso
È una malattia genetica rara che si manifesta con episodi ricorrenti di edema. Nella maggior parte dei casi è causato da un deficit congenito di sintesi o di funzionalità del C1 inibitore (C1-INH), ma esistono altre mutazioni che causano angioedema ereditario con livelli di C1-INH normali e che hanno una frequenza molto ridotta.
L’HAE è caratterizzato da una reazione vascolare transitoria dei tessuti profondi del derma, del sottocute, di mucosa o di sottomucosa, che causa aumentata permeabilità localizzata dei vasi sanguigni con conseguente stravaso di essudato plasmatico e rigonfiamento dei tessuti.
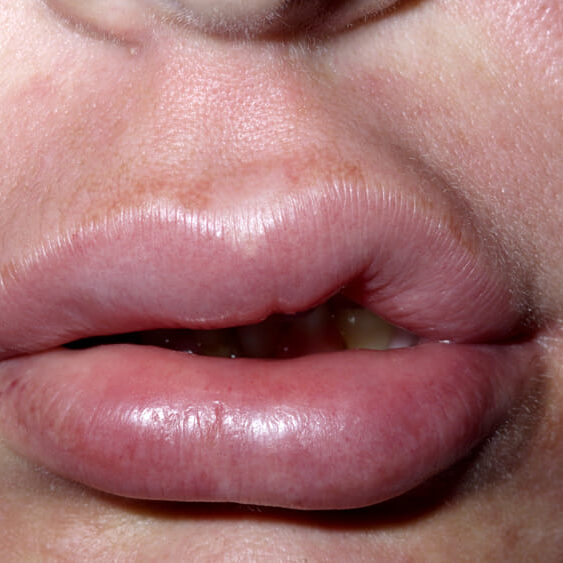
Gli edemi della cute possono rendere più difficoltose le attività quotidiane e provocare dolore, mentre quelli del viso possono alterare la fisionomia e causare difficoltà nel parlare o nel mangiare/bere. L’edema della parete gastrointestinale può causare sensazione di gonfiore e dolori addominali simili ad una colica, ed eventualmente essere associato a vomito e/o diarrea. L’edema della glottide è una vera emergenza medica in quanto può determinare l’ostruzione completa delle alte vie aeree respiratorie e causare morte per asfissia.
Gli attacchi di HAE durano in media 48-72 ore, finanche 5 giorni, e sono autolimitanti. I sintomi possono comparire dopo traumi, infezioni, procedure mediche o stress.
Sedi
Cute, viso (in particolare palpebre, labbra e lingua), organi addominali (in particolare intestino tenue e grasso), alte vie respiratorie, gonadi (in particolare testicoli).
Diagnosi differenziale
Angioedema bradichinina indotto: angioedema acquisito con carenza di C1-INH; angioedema causato da ACE-inibitori.
Angioedema indotto da mediatori dei mastociti: angioedema con anafilassi, angioedema da farmaci, angioedema IgE mediato con o senza orticaria, angioedema idiopatico acquisito.
Terapia
In acuto: C1-INH derivato dal plasma; C1-INH ricombinante umano; ecallantide (inibitore della callicreina); icatibant (antagonista del recettore B2 della bradichinina); plasma fresco congelato; acido tranexamico.
Profilassi a breve termine: nei pazienti in profilassi a lungo termine, viene adattato lo schema di somministrazione con una dose appena prima del possibile evento scatenante (es intervento); 5 giorni di androgeni prima e 2 dopo l’evento; acido tranexanico.
Profilassi a lungo termine: Lanadelumab, anticoro monoclonale inibitore plasmatico della callicreina; C1-INH derivato dal plasma ev o sc; androgeni; acido tranexanico.